
Giriş Yap
Genetic Disorder Causing Progressive Motor and Cognitive Decline in Middle-Aged Woman
Temel Kavramlar
Huntington's disease, an inherited neurodegenerative disorder, causes progressive motor, cognitive, and psychiatric symptoms in a 51-year-old woman, requiring comprehensive clinical evaluation and genetic testing for diagnosis and management.
Özet
The content describes the case of a 51-year-old woman presenting with worsening short-term memory, difficulty with organization and planning, and involuntary movements (chorea) in her extremities. The patient has a family history of dementia and balance problems accompanied by depression and temper outbursts.
Based on the patient's symptoms, family history, neuroimaging findings, and genetic testing, she was diagnosed with Huntington's disease (HD), an inherited, autosomal dominant, progressive, and fatal neurodegenerative disorder. HD is characterized by involuntary movements, behavioral impairment, and cognitive decline, typically affecting patients between the ages of 30 and 50.
The differential diagnosis considered other neurological conditions, including multiple sclerosis, neuroacanthocytosis, systemic lupus erythematosus, Alzheimer's disease, frontotemporal dementia, corticobasal degeneration, Parkinson's disease, and various spinocerebellar ataxias. However, the patient's presentation, family history, and genetic testing results confirmed the diagnosis of HD.
HD is caused by an expanded CAG (cytosine-adenine-guanine) repeat in the HTT gene, which codes for an abnormal version of the huntingtin protein. The number of CAG repeats determines the risk and severity of the disease, with more than 40 repeats being diagnostic for HD.
The content outlines the management of HD, which includes symptomatic treatments for chorea, behavioral and psychiatric symptoms, cognitive impairment, and seizures (in juvenile cases). Supportive care, physical and occupational therapy, and patient/family education are also crucial. Investigational therapies targeting the underlying genetic cause are being studied, but no effective treatment for preventing or reversing the progression of HD is currently available.
HD is a relentlessly progressive disorder, leading to disability and death, with a mean survival of 15-20 years from the onset of symptoms. Early diagnosis is important to provide genetic counseling, access to symptom-relieving treatments, and support for patients and their families.
Progressive Motor, Cognitive Decline in 51-Year-Old Woman
İstatistikler
The patient had a 43-repeat CAG expansion of the HTT gene, which is diagnostic for Huntington's disease.
Brain MRI showed striatal atrophy in the caudate nucleus.
The mean age at death in major HD studies ranged from 51 to 57 years, with a mean duration of illness of approximately 19 years.
Alıntılar
"HD is an inherited, autosomal dominant, relentlessly progressive, fatal neurodegenerative disease characterized by involuntary movements, behavioral impairment, and cognitive decline."
"The number of CAG repeats present in the HTT gene determines whether an individual will have HD. Those with six to 35 CAG repeats will be unaffected (although, as noted above, more than 26 copies is considered abnormal; such individuals may pass on a longer expansion to any offspring)."
"HD is a relentlessly progressive disorder leading to disability and death. The mean age at death in all major series ranged from 51 to 57 years, but the real-world range is broader. Duration of illness varies considerably, with a mean of approximately 19 years."
Önemli Bilgiler Şuradan Elde Edildi
by Karen E. And... : www.medscape.com 05-01-2024
https://www.medscape.com/viewarticle/dont-miss-dx-progressive-motor-behavioral-and-cognitive-2024a10008d5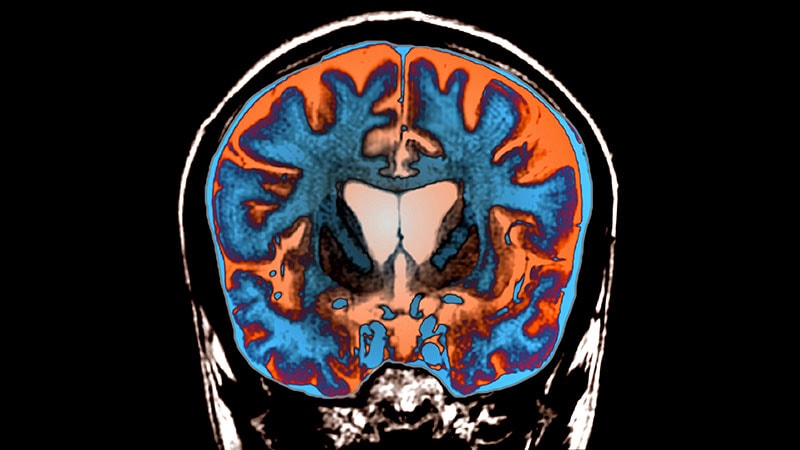
Daha Derin Sorular
What are the potential genetic and environmental factors that may influence the age of onset and progression of Huntington's disease?
Huntington's disease (HD) is primarily caused by an expanded CAG repeat in the HTT gene, leading to the production of a mutant huntingtin protein. The number of CAG repeats in the gene is a major genetic factor influencing the age of onset and progression of HD. Individuals with 40 or more CAG repeats typically manifest the disease, with earlier onset and more severe symptoms associated with higher repeat numbers. Additionally, genetic modifiers and epigenetic factors can influence the variability in age of onset and disease progression among individuals with HD.
Environmental factors can also play a role in the age of onset and progression of HD. Lifestyle factors such as diet, exercise, and exposure to toxins may impact the rate of disease progression. Stress and other environmental stressors can exacerbate symptoms and affect the overall well-being of individuals with HD. However, the extent to which environmental factors contribute to the variability in age of onset and progression of HD is not fully understood and requires further research.
How do the cognitive and psychiatric symptoms of Huntington's disease differ from other neurodegenerative disorders, and what are the implications for patient management and caregiver support?
Cognitive and psychiatric symptoms in Huntington's disease (HD) are distinct from other neurodegenerative disorders in several ways. HD is characterized by a triad of symptoms: motor dysfunction, cognitive decline, and psychiatric disturbances. Cognitive impairment in HD typically involves deficits in executive function, attention, and working memory, leading to difficulties in planning, organizing, and decision-making. Psychiatric symptoms include depression, anxiety, irritability, and apathy, which can significantly impact the quality of life for patients with HD.
In comparison to other neurodegenerative disorders like Alzheimer's disease, where memory impairment is a hallmark feature, HD presents with a unique profile of cognitive and psychiatric symptoms. The management of cognitive and psychiatric symptoms in HD requires a multidisciplinary approach involving pharmacological interventions, psychotherapy, and supportive care. Caregiver support is crucial in addressing the complex needs of patients with HD, as they may require assistance with daily activities, emotional support, and access to community resources. Education and training for caregivers on how to manage behavioral changes and provide a safe and nurturing environment for patients with HD are essential for optimal patient outcomes.
Given the autosomal dominant inheritance pattern of Huntington's disease, what are the ethical considerations and challenges in genetic testing and counseling for at-risk individuals, especially those who are asymptomatic?
Genetic testing and counseling for individuals at risk of Huntington's disease (HD) present several ethical considerations and challenges due to the autosomal dominant inheritance pattern of the disease. One of the primary ethical considerations is the potential psychological impact of genetic testing on at-risk individuals, especially those who are asymptomatic. The knowledge of carrying the mutated allele can lead to anxiety, depression, and uncertainty about the future, affecting the mental well-being of individuals and their families.
Another ethical challenge is the issue of genetic discrimination, where individuals may face discrimination in employment, insurance, or social settings based on their genetic status. Privacy concerns regarding the disclosure of genetic information and the implications for family members who may also be at risk of HD are important ethical considerations in genetic testing and counseling.
Furthermore, the decision-making process for at-risk individuals regarding genetic testing can be complex, involving considerations of autonomy, beneficence, and non-maleficence. Genetic counselors play a crucial role in providing information, support, and guidance to individuals and families facing decisions about genetic testing for HD. Respect for autonomy, informed consent, and confidentiality are key ethical principles that should guide the genetic counseling process for at-risk individuals, ensuring that they have the necessary information and support to make informed decisions about testing and future planning.
0
Bu Sayfayı Görselleştir
Tespit Edilemeyen AI ile Oluştur
Başka Bir Dile Çevir
Akademik Arama
Hakkında
Ürünler | Kaynaklar
© 2024 by Linnk AI